2. scVelo: Dynamical Modeling¶
https://scvelo.readthedocs.io/DynamicalModeling.html
最終更新日: 2023/10/1
ここでは全体的な転写ダイナミクスを求めるために一般化力学モデル (generalized dynamical model) を用います。
これにより、潜在時間や潜在的なドライバー遺伝子の同定など、いくつかの付加的な知見が得られます。
前回のチュートリアルと同様に、pancreasの内分泌系の発生をデータに用います。
[2]:
import scvelo as scv
scv.logging.print_version()
scv.settings.verbosity = 3 # show errors(0), warnings(1), info(2), hints(3)
scv.settings.presenter_view = True # set max width size for presenter view
scv.settings.set_figure_params('scvelo') # for beautified visualization
Running scvelo 0.2.5 (python 3.8.16) on 2023-10-01 18:11.
ERROR: XMLRPC request failed [code: -32500]
RuntimeError: PyPI no longer supports 'pip search' (or XML-RPC search). Please use https://pypi.org/search (via a browser) instead. See https://warehouse.pypa.io/api-reference/xml-rpc.html#deprecated-methods for more information.
2.1. データの準備¶
前準備として、HVGの選択、総サイズによる遺伝子発現量の正規化・対数化、速度推定のためのモーメント計算を実行します。 詳しい説明は前のチュートリアルを参照。
[3]:
adata = scv.datasets.pancreas()
scv.pp.filter_and_normalize(adata, min_shared_counts=20, n_top_genes=2000)
scv.pp.moments(adata, n_pcs=30, n_neighbors=30)
Filtered out 20801 genes that are detected 20 counts (shared).
Normalized count data: X, spliced, unspliced.
Extracted 2000 highly variable genes.
Logarithmized X.
computing neighbors
finished (0:00:06) --> added
'distances' and 'connectivities', weighted adjacency matrices (adata.obsp)
computing moments based on connectivities
finished (0:00:00) --> added
'Ms' and 'Mu', moments of un/spliced abundances (adata.layers)
2.2. Dynamical Model¶
スプライシング動態の完全な転写ダイナミクスを学習するために力学モデルを実行する。
このモデルは尤度ベースの期待値最大化フレームワークでを用いて、反応速度と細胞固有の潜在変数、すなわち転写状態と細胞内潜在時間のパラメータを反復的に推定することで求められます。これにより、各遺伝子のスプライスされていない/スプライスされた位相の軌跡を学習します。
[4]:
scv.tl.recover_dynamics(adata)
scv.tl.velocity(adata, mode='dynamical')
scv.tl.velocity_graph(adata)
recovering dynamics (using 1/40 cores)
finished (0:10:52) --> added
'fit_pars', fitted parameters for splicing dynamics (adata.var)
computing velocities
finished (0:00:04) --> added
'velocity', velocity vectors for each individual cell (adata.layers)
computing velocity graph (using 1/40 cores)
finished (0:00:10) --> added
'velocity_graph', sparse matrix with cosine correlations (adata.uns)
動的モデルの実行には時間がかかります。そこで、adata.write('data/pancreas.h5ad')で結果を保存し、後でadata = scv.read('data/pancreas.h5ad')で読み込むようにします。
[5]:
scv.pl.velocity_embedding_stream(adata, basis='umap')
computing velocity embedding
finished (0:00:00) --> added
'velocity_umap', embedded velocity vectors (adata.obsm)
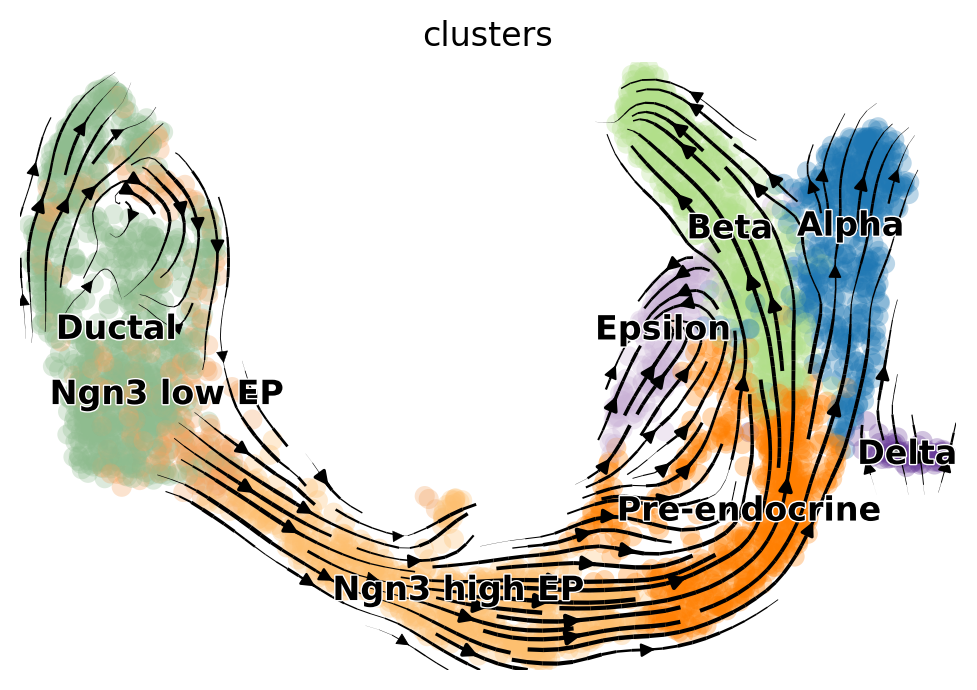
2.3. Kinetic rate paramters¶
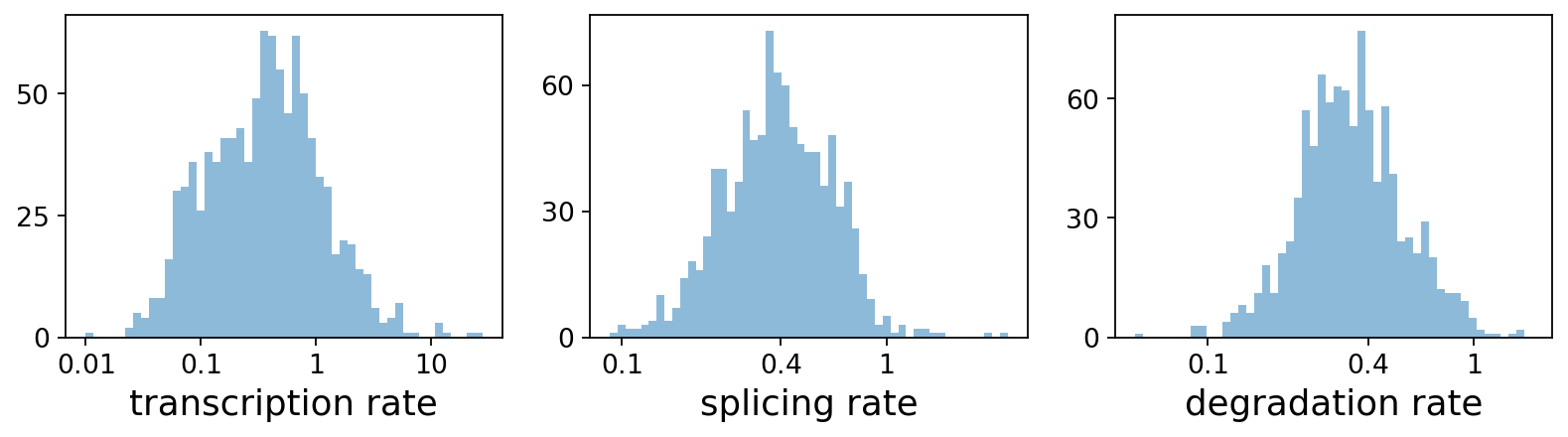
RNAの転写、スプライシング、分解の速度は、実験データを必要とせずに推定できます。 これらは、細胞の同一性や表現型の不均一性をよりよく理解するために有用です。
[7]:
df = adata.var
df = df[(df['fit_likelihood'] > .1) & df['velocity_genes'] == True]
kwargs = dict(xscale='log', fontsize=16)
with scv.GridSpec(ncols=3) as pl:
pl.hist(df['fit_alpha'], xlabel='transcription rate', **kwargs)
pl.hist(df['fit_beta'] * df['fit_scaling'], xlabel='splicing rate', xticks=[.1, .4, 1], **kwargs)
pl.hist(df['fit_gamma'], xlabel='degradation rate', xticks=[.1, .4, 1], **kwargs)
scv.get_df(adata, 'fit*', dropna=True).head()
[7]:
| fit_r2 | fit_alpha | fit_beta | fit_gamma | fit_t_ | fit_scaling | fit_std_u | fit_std_s | fit_likelihood | fit_u0 | fit_s0 | fit_pval_steady | fit_steady_u | fit_steady_s | fit_variance | fit_alignment_scaling | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| index | ||||||||||||||||
| Sntg1 | 0.401981 | 0.015726 | 0.005592 | 0.088792 | 23.404254 | 42.849447 | 1.029644 | 0.030838 | 0.406523 | 0.0 | 0.0 | 0.159472 | 2.470675 | 0.094304 | 0.149138 | 5.355590 |
| Snhg6 | 0.125072 | 0.389126 | 2.981982 | 0.260322 | 6.981600 | 0.070368 | 0.037113 | 0.245248 | 0.243441 | 0.0 | 0.0 | 0.403409 | 0.106128 | 0.596630 | 0.762252 | 2.037296 |
| Sbspon | 0.624803 | 0.464865 | 2.437113 | 0.379645 | 3.785993 | 0.154771 | 0.058587 | 0.178859 | 0.252135 | 0.0 | 0.0 | 0.182088 | 0.164805 | 0.430623 | 0.674312 | 1.193015 |
| Mcm3 | 0.292389 | 3.096367 | 39.995796 | 0.638543 | 2.049463 | 0.013943 | 0.016253 | 0.673142 | 0.228207 | 0.0 | 0.0 | 0.467683 | 0.051432 | 1.927742 | 0.687468 | 0.887607 |
| Fam135a | 0.384662 | 0.172335 | 0.118088 | 0.204538 | 11.239574 | 1.124040 | 0.356525 | 0.149868 | 0.283343 | 0.0 | 0.0 | 0.387921 | 1.345830 | 0.393197 | 0.671096 | 3.390194 |
推定される遺伝子特異的パラメータ群は以下の通りです: - 転写率(fit_alpha) - スプライシング(fit_beta) - 分解(fit_gamma) - スイッチングタイムポイント(fit_t_) - 過少に代表される未スプライスリードを調整するためのスケーリングパラメータ(fit_scaling) - 未スプライスリードとスプライスリードの標準偏差(fit_std_u、fit_std_s) - 遺伝子尤度(fit_likelihood) - 推論された定常状態レベル(fit_steady_u, fit_steady_s) - それに対応する
p値(fit_pval_steady_u, fit_pval_steady_s) - モデル全体の分散(fit_variance) - 遺伝子ごとの潜在時間を普遍的な遺伝子共有潜 在時間に揃えるためのスケーリング係数(fit_alignment_scaling)。
2.4. 潜在時間¶
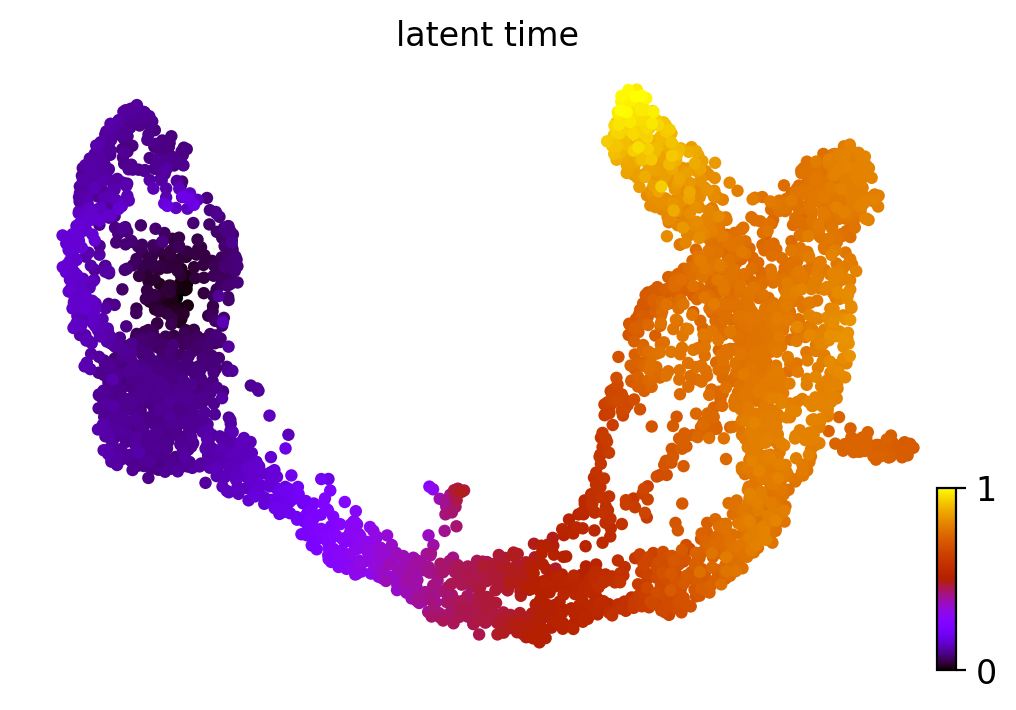
力学的モデルは、基礎となる細胞プロセスの潜在時間を復元します。この潜伏時間は細胞の内部時計を表し、細胞が分化する際に経験する実時間を転写ダイナミクスのみに基づいて近似します。
[8]:
scv.tl.latent_time(adata)
scv.pl.scatter(adata, color='latent_time', color_map='gnuplot', size=80)
computing terminal states
identified 2 regions of root cells and 1 region of end points .
finished (0:00:00) --> added
'root_cells', root cells of Markov diffusion process (adata.obs)
'end_points', end points of Markov diffusion process (adata.obs)
computing latent time using root_cells as prior
finished (0:00:01) --> added
'latent_time', shared time (adata.obs)
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scvelo/plotting/utils.py:869: MatplotlibDeprecationWarning: The draw_all function was deprecated in Matplotlib 3.6 and will be removed two minor releases later. Use fig.draw_without_rendering() instead.
cb.draw_all()
[9]:
top_genes = adata.var['fit_likelihood'].sort_values(ascending=False).index[:300]
scv.pl.heatmap(adata, var_names=top_genes, sortby='latent_time', col_color='clusters', n_convolve=100)
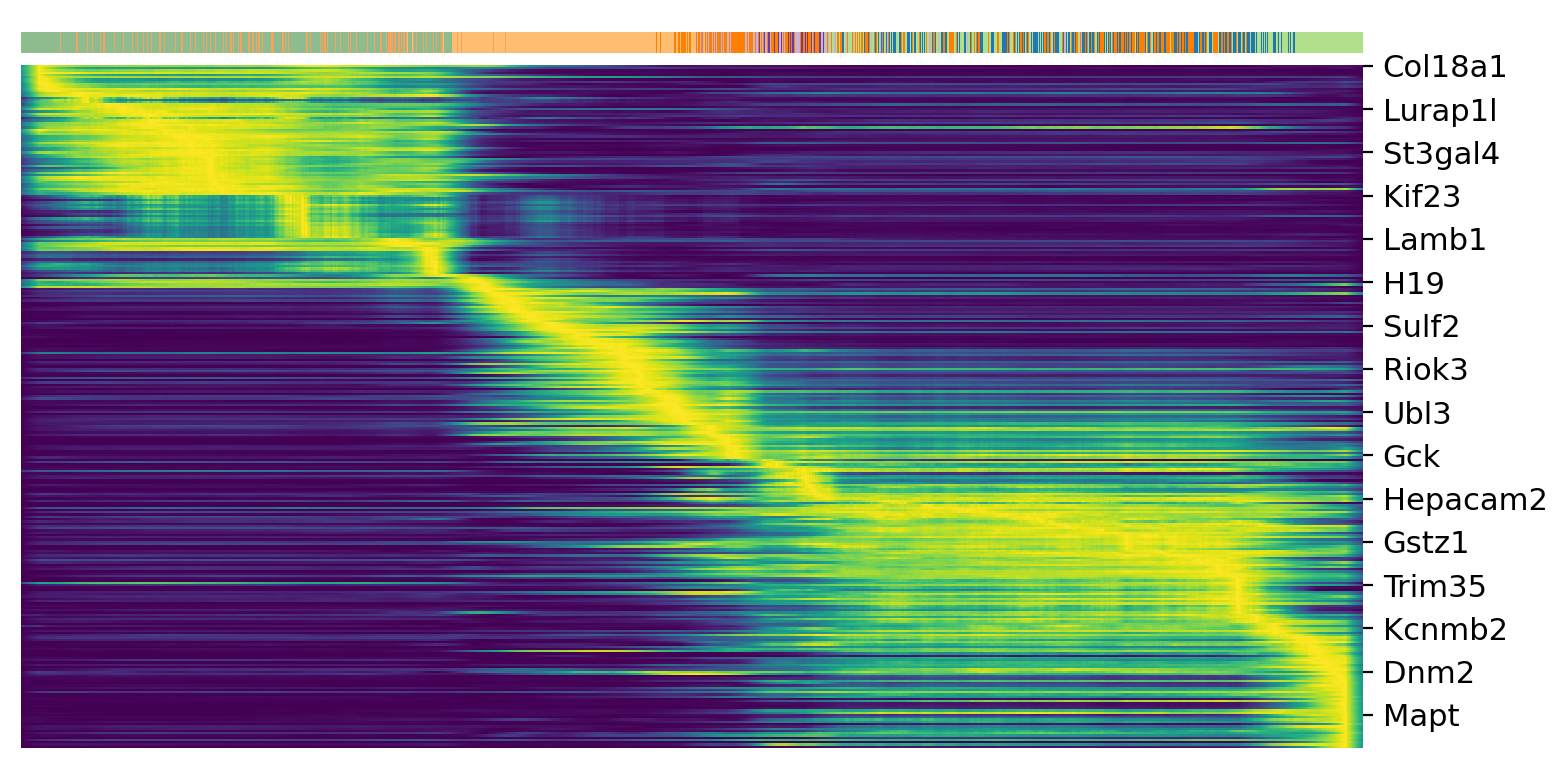
2.5. Top-likelihood genes¶
ドライバー遺伝子は顕著な動的挙動を示し、動的モデルでの高い尤度による特徴付けによって、系統的に検出されます。
[10]:
top_genes = adata.var['fit_likelihood'].sort_values(ascending=False).index
scv.pl.scatter(adata, basis=top_genes[:15], ncols=5, frameon=False)

[11]:
var_names = ['Actn4', 'Ppp3ca', 'Cpe', 'Nnat']
scv.pl.scatter(adata, var_names, frameon=False)
scv.pl.scatter(adata, x='latent_time', y=var_names, frameon=False)
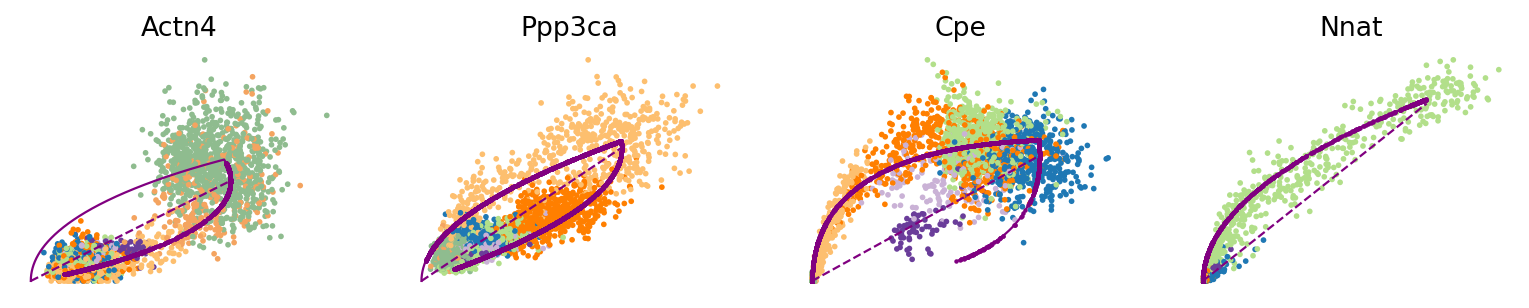
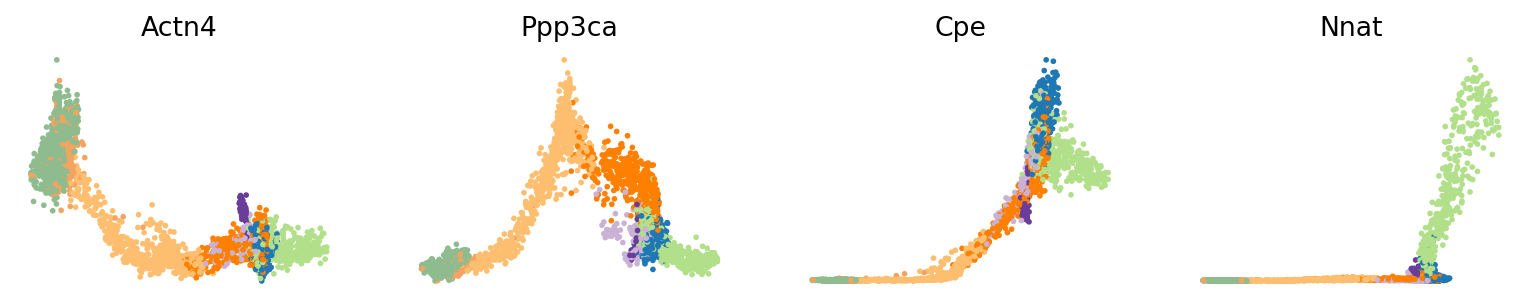
2.6. Cluster-specific top-likelihood genes¶
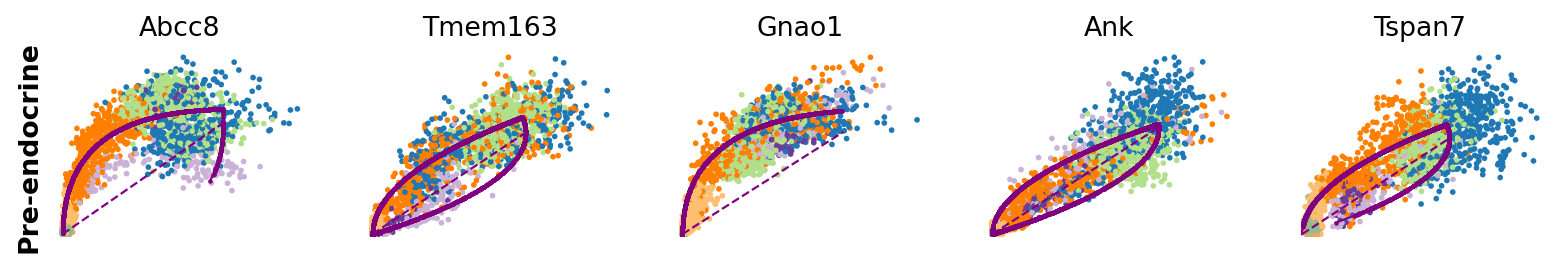
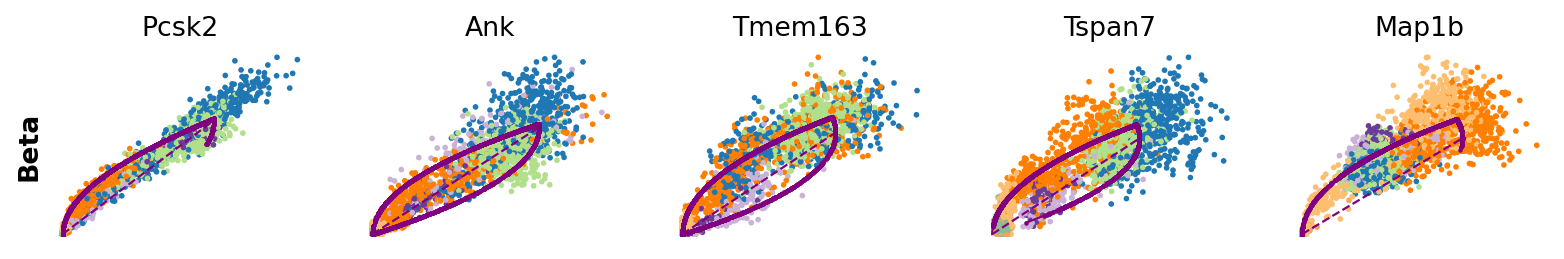
さらに、部分的な遺伝子の尤度を細胞の各クラスタに対して計算することで、潜在的なドライバーをクラスタごとに特定することができます。
[12]:
scv.tl.rank_dynamical_genes(adata, groupby='clusters')
df = scv.get_df(adata, 'rank_dynamical_genes/names')
df.head(5)
ranking genes by cluster-specific likelihoods
finished (0:00:03) --> added
'rank_dynamical_genes', sorted scores by group ids (adata.uns)
[12]:
| Ductal | Ngn3 low EP | Ngn3 high EP | Pre-endocrine | Beta | Alpha | Delta | Epsilon | |
|---|---|---|---|---|---|---|---|---|
| 0 | Dcdc2a | Dcdc2a | Rbfox3 | Abcc8 | Pcsk2 | Cpe | Pcsk2 | Tox3 |
| 1 | Top2a | Adk | Mapre3 | Tmem163 | Ank | Gnao1 | Rap1b | Rnf130 |
| 2 | Nfib | Mki67 | Btbd17 | Gnao1 | Tmem163 | Pak3 | Pak3 | Meis2 |
| 3 | Wfdc15b | Rap1gap2 | Sulf2 | Ank | Tspan7 | Pim2 | Abcc8 | Adk |
| 4 | Cdk1 | Top2a | Tcp11 | Tspan7 | Map1b | Map1b | Klhl32 | Rap1gap2 |
[13]:
for cluster in ['Ductal', 'Ngn3 high EP', 'Pre-endocrine', 'Beta']:
scv.pl.scatter(adata, df[cluster][:5], ylabel=cluster, frameon=False)
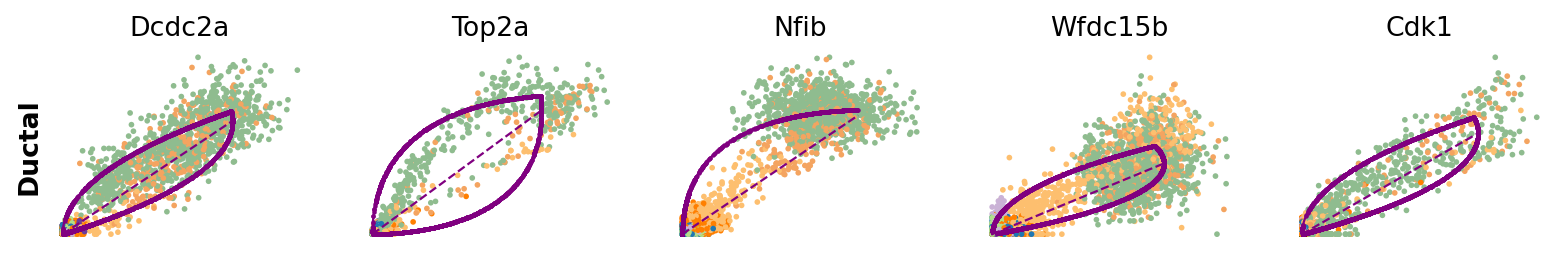
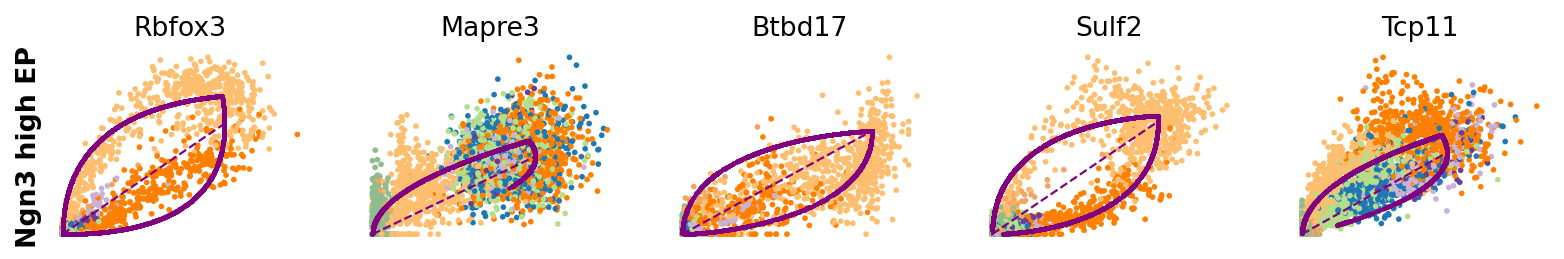
[14]:
import session_info
session_info.show()
[14]:
Click to view session information
----- anndata 0.9.1 pandas 1.5.3 scvelo 0.2.5 session_info 1.0.0 -----
Click to view modules imported as dependencies
PIL 9.4.0 asciitree NA asttokens NA backcall 0.2.0 beta_ufunc NA binom_ufunc NA bottleneck 1.3.5 cffi 1.15.1 cloudpickle 2.2.1 colorama 0.4.6 comm 0.1.2 cycler 0.10.0 cython_runtime NA dask 2023.5.0 dateutil 2.8.2 debugpy 1.5.1 decorator 5.1.1 defusedxml 0.7.1 entrypoints 0.4 executing 0.8.3 fasteners 0.18 h5py 3.8.0 hypergeom_ufunc NA igraph 0.10.4 importlib_metadata NA importlib_resources NA ipykernel 6.19.2 ipython_genutils 0.2.0 ipywidgets 8.0.4 jedi 0.18.1 jinja2 3.1.2 joblib 1.2.0 jupyter_server 1.23.4 kiwisolver 1.4.4 leidenalg 0.9.1 llvmlite 0.39.1 louvain 0.8.0 markupsafe 2.1.1 matplotlib 3.7.1 matplotlib_inline 0.1.6 mpl_toolkits NA msgpack 1.0.5 natsort 8.3.1 nbinom_ufunc NA ncf_ufunc NA numba 0.56.4 numcodecs 0.11.0 numexpr 2.8.4 numpy 1.23.5 packaging 23.0 parso 0.8.3 patsy 0.5.3 pexpect 4.8.0 pickleshare 0.7.5 pkg_resources NA platformdirs 2.5.2 plotly 5.14.1 prompt_toolkit 3.0.36 psutil 5.9.0 ptyprocess 0.7.0 pure_eval 0.2.2 pycparser 2.21 pydev_ipython NA pydevconsole NA pydevd 2.6.0 pydevd_concurrency_analyser NA pydevd_file_utils NA pydevd_plugins NA pydevd_tracing NA pygments 2.15.1 pynndescent 0.5.10 pyparsing 3.0.9 pytz 2022.7 scanpy 1.9.3 scipy 1.9.1 seaborn 0.12.2 setuptools 66.0.0 six 1.16.0 sklearn 1.2.2 stack_data 0.2.0 statsmodels 0.14.0 texttable 1.6.7 threadpoolctl 3.1.0 tlz 0.12.0 toolz 0.12.0 tornado 6.2 tqdm 4.65.0 traitlets 5.7.1 typing_extensions NA umap 0.5.3 wcwidth 0.2.5 yaml 6.0 zarr 2.14.2 zipp NA zmq 25.0.2 zope NA
----- IPython 8.12.0 jupyter_client 8.1.0 jupyter_core 5.3.0 jupyterlab 3.5.3 notebook 6.5.4 ----- Python 3.8.16 (default, Mar 2 2023, 03:21:46) [GCC 11.2.0] Linux-5.15.0-79-generic-x86_64-with-glibc2.17 ----- Session information updated at 2023-10-01 18:56