1. Scanpy: Preprocessing and clustering 3k PBMCs¶
https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html
(更新日:2024-2-6 with ShortCake v1.9.1)
Scanpyを用いたクラスタリング解析の基本的なワークフローを紹介します。 Google ColabまたはJupyter notebook上で作業を行います。 内容はSeuratのGuided tutorialとほぼ同じですので、そちらもあわせて参考にしてください。
1.1. 前準備¶
Google Colabで本チュートリアルを実行する場合は まず下記コマンドでScanpyをインストールしてください。
[ ]:
!pip install seaborn scikit-learn statsmodels numba python-igraph louvain leidenalg scanpy
Requirement already satisfied: seaborn in /usr/local/lib/python3.7/dist-packages (0.11.1)
Requirement already satisfied: scikit-learn in /usr/local/lib/python3.7/dist-packages (0.22.2.post1)
Requirement already satisfied: statsmodels in /usr/local/lib/python3.7/dist-packages (0.10.2)
Requirement already satisfied: numba in /usr/local/lib/python3.7/dist-packages (0.51.2)
Requirement already satisfied: python-igraph in /usr/local/lib/python3.7/dist-packages (0.9.1)
Requirement already satisfied: louvain in /usr/local/lib/python3.7/dist-packages (0.7.0)
Requirement already satisfied: leidenalg in /usr/local/lib/python3.7/dist-packages (0.8.4)
Requirement already satisfied: scanpy in /usr/local/lib/python3.7/dist-packages (1.7.2)
Requirement already satisfied: pandas>=0.23 in /usr/local/lib/python3.7/dist-packages (from seaborn) (1.1.5)
Requirement already satisfied: matplotlib>=2.2 in /usr/local/lib/python3.7/dist-packages (from seaborn) (3.2.2)
Requirement already satisfied: scipy>=1.0 in /usr/local/lib/python3.7/dist-packages (from seaborn) (1.4.1)
Requirement already satisfied: numpy>=1.15 in /usr/local/lib/python3.7/dist-packages (from seaborn) (1.19.5)
Requirement already satisfied: joblib>=0.11 in /usr/local/lib/python3.7/dist-packages (from scikit-learn) (1.0.1)
Requirement already satisfied: patsy>=0.4.0 in /usr/local/lib/python3.7/dist-packages (from statsmodels) (0.5.1)
Requirement already satisfied: setuptools in /usr/local/lib/python3.7/dist-packages (from numba) (56.0.0)
Requirement already satisfied: llvmlite<0.35,>=0.34.0.dev0 in /usr/local/lib/python3.7/dist-packages (from numba) (0.34.0)
Requirement already satisfied: texttable>=1.6.2 in /usr/local/lib/python3.7/dist-packages (from python-igraph) (1.6.3)
Requirement already satisfied: tqdm in /usr/local/lib/python3.7/dist-packages (from scanpy) (4.41.1)
Requirement already satisfied: umap-learn>=0.3.10 in /usr/local/lib/python3.7/dist-packages (from scanpy) (0.5.1)
Requirement already satisfied: sinfo in /usr/local/lib/python3.7/dist-packages (from scanpy) (0.3.1)
Requirement already satisfied: networkx>=2.3 in /usr/local/lib/python3.7/dist-packages (from scanpy) (2.5.1)
Requirement already satisfied: legacy-api-wrap in /usr/local/lib/python3.7/dist-packages (from scanpy) (1.2)
Requirement already satisfied: importlib-metadata>=0.7; python_version < "3.8" in /usr/local/lib/python3.7/dist-packages (from scanpy) (3.10.1)
Requirement already satisfied: h5py>=2.10.0 in /usr/local/lib/python3.7/dist-packages (from scanpy) (2.10.0)
Requirement already satisfied: tables in /usr/local/lib/python3.7/dist-packages (from scanpy) (3.4.4)
Requirement already satisfied: packaging in /usr/local/lib/python3.7/dist-packages (from scanpy) (20.9)
Requirement already satisfied: natsort in /usr/local/lib/python3.7/dist-packages (from scanpy) (5.5.0)
Requirement already satisfied: anndata>=0.7.4 in /usr/local/lib/python3.7/dist-packages (from scanpy) (0.7.6)
Requirement already satisfied: python-dateutil>=2.7.3 in /usr/local/lib/python3.7/dist-packages (from pandas>=0.23->seaborn) (2.8.1)
Requirement already satisfied: pytz>=2017.2 in /usr/local/lib/python3.7/dist-packages (from pandas>=0.23->seaborn) (2018.9)
Requirement already satisfied: cycler>=0.10 in /usr/local/lib/python3.7/dist-packages (from matplotlib>=2.2->seaborn) (0.10.0)
Requirement already satisfied: kiwisolver>=1.0.1 in /usr/local/lib/python3.7/dist-packages (from matplotlib>=2.2->seaborn) (1.3.1)
Requirement already satisfied: pyparsing!=2.0.4,!=2.1.2,!=2.1.6,>=2.0.1 in /usr/local/lib/python3.7/dist-packages (from matplotlib>=2.2->seaborn) (2.4.7)
Requirement already satisfied: six in /usr/local/lib/python3.7/dist-packages (from patsy>=0.4.0->statsmodels) (1.15.0)
Requirement already satisfied: pynndescent>=0.5 in /usr/local/lib/python3.7/dist-packages (from umap-learn>=0.3.10->scanpy) (0.5.2)
Requirement already satisfied: stdlib-list in /usr/local/lib/python3.7/dist-packages (from sinfo->scanpy) (0.8.0)
Requirement already satisfied: decorator<5,>=4.3 in /usr/local/lib/python3.7/dist-packages (from networkx>=2.3->scanpy) (4.4.2)
Requirement already satisfied: get-version>=2.0.4 in /usr/local/lib/python3.7/dist-packages (from legacy-api-wrap->scanpy) (2.2)
Requirement already satisfied: typing-extensions>=3.6.4; python_version < "3.8" in /usr/local/lib/python3.7/dist-packages (from importlib-metadata>=0.7; python_version < "3.8"->scanpy) (3.7.4.3)
Requirement already satisfied: zipp>=0.5 in /usr/local/lib/python3.7/dist-packages (from importlib-metadata>=0.7; python_version < "3.8"->scanpy) (3.4.1)
Requirement already satisfied: numexpr>=2.5.2 in /usr/local/lib/python3.7/dist-packages (from tables->scanpy) (2.7.3)
Requirement already satisfied: xlrd<2.0 in /usr/local/lib/python3.7/dist-packages (from anndata>=0.7.4->scanpy) (1.1.0)
1.1.1. データダウンロード(初回のみ)¶
Jupyterでは冒頭に ! 記号をつけるとLinuxコマンドを実行することができます。これをマジックコマンドと言います。
mkdirはディレクトリを新規作成するLinuxコマンドです。
wgetを使って、今回の解析に用いるデータをWebからダウンロードし、ローカルに保存します。データサイズは7.27Mです。
tarコマンドでダウンロードされた圧縮ファイルを解凍します。
[1]:
!mkdir -p data write
!wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz
!cd data; tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz
--2024-02-06 19:27:21-- http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
Resolving cf.10xgenomics.com (cf.10xgenomics.com)... 104.18.0.173, 104.18.1.173, 2606:4700::6812:ad, ...
Connecting to cf.10xgenomics.com (cf.10xgenomics.com)|104.18.0.173|:80... connected.
HTTP request sent, awaiting response... 301 Moved Permanently
Location: https://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz [following]
--2024-02-06 19:27:21-- https://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
Connecting to cf.10xgenomics.com (cf.10xgenomics.com)|104.18.0.173|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 7621991 (7.3M) [application/x-tar]
Saving to: ‘data/pbmc3k_filtered_gene_bc_matrices.tar.gz’
data/pbmc3k_filtere 100%[===================>] 7.27M 18.4MB/s in 0.4s
2024-02-06 19:27:21 (18.4 MB/s) - ‘data/pbmc3k_filtered_gene_bc_matrices.tar.gz’ saved [7621991/7621991]
[118]:
# ライブラリ読み込み
import numpy as np
import pandas as pd
import scanpy as sc
# ログなどのパラメータ設定
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.settings.set_figure_params(dpi=80, facecolor='white')
results_file = 'write/pbmc3k.h5ad' # 出力ファイル名
入力データ(10X CellRangerで生成されたディレクトリ)を読み込み、AnnDataオブジェクト に格納
[119]:
adata = sc.read_10x_mtx(
'data/filtered_gene_bc_matrices/hg19/', # 入力ディレクトリ
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
cache=True) # 計算の高速化のためにキャッシュを利用する
adata.var_names_make_unique() # this is unnecessary if using `var_names='gene_ids'` in `sc.read_10x_mtx`
adata
... reading from cache file cache/data-filtered_gene_bc_matrices-hg19-matrix.h5ad
[119]:
AnnData object with n_obs × n_vars = 2700 × 32738
var: 'gene_ids'
2700細胞 x 32738遺伝子のAnnDataが作成されました。
1.2. Preprocessing¶
各細胞の全リードに対して占める割合が最も高い遺伝子を表示します。
[120]:
sc.pl.highest_expr_genes(adata, n_top=20, )
normalizing counts per cell
finished (0:00:00)
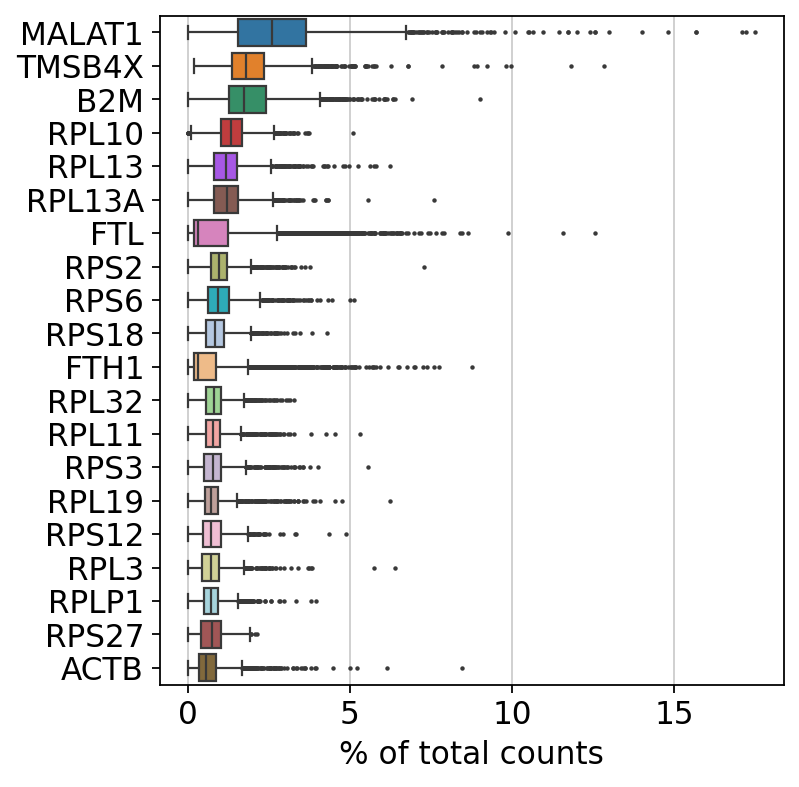
1.2.1. フィルタリング¶
発現遺伝子数が200未満の細胞、発現細胞数が3未満の遺伝子をフィルタします。
[121]:
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
filtered out 19024 genes that are detected in less than 3 cells
発現細胞数が3未満の19,024遺伝子がフィルタされ、2700細胞 x 13714遺伝子のAnnDataになりました。
[122]:
adata
[122]:
AnnData object with n_obs × n_vars = 2700 × 13714
obs: 'n_genes'
var: 'gene_ids', 'n_cells'
次に、品質評価に重要なミトコンドリア遺伝子の発現量を計測します。 ミトコンドリアの発現割合が高い細胞は死細胞とみなされます。 (Lun, McCarthy & Marioni, 2017)
pp.calculate_qc_metrics 関数によって多数の品質評価指標を取得できます。
[123]:
adata.var['mt'] = adata.var_names.str.startswith('MT-')
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
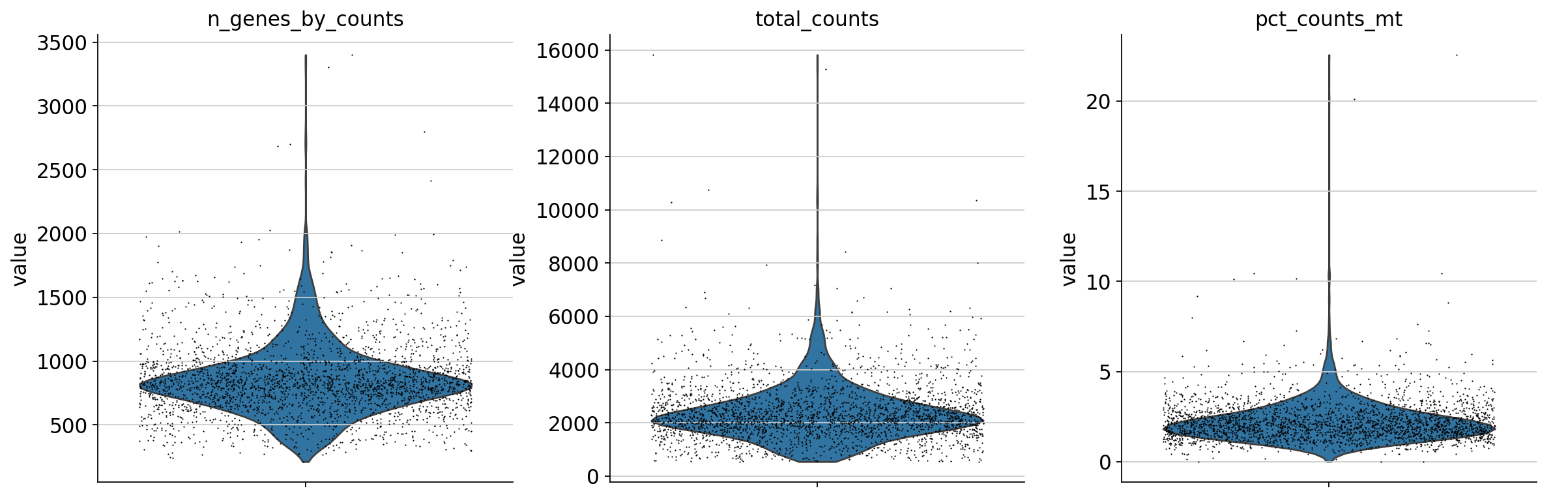
violin plotで以下の尺度を評価します。 - カウントマトリックス中の発現遺伝子数 - 細胞あたりの総カウント数 - ミトコンドリア遺伝子におけるカウントの割合
[124]:
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt'],
jitter=0.4, multi_panel=True)
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/seaborn/axisgrid.py:123: UserWarning: The figure layout has changed to tight
self._figure.tight_layout(*args, **kwargs)
[125]:
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt')
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')
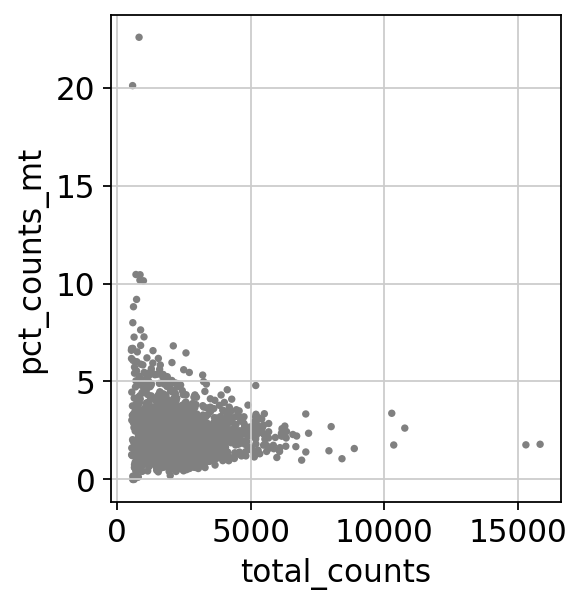
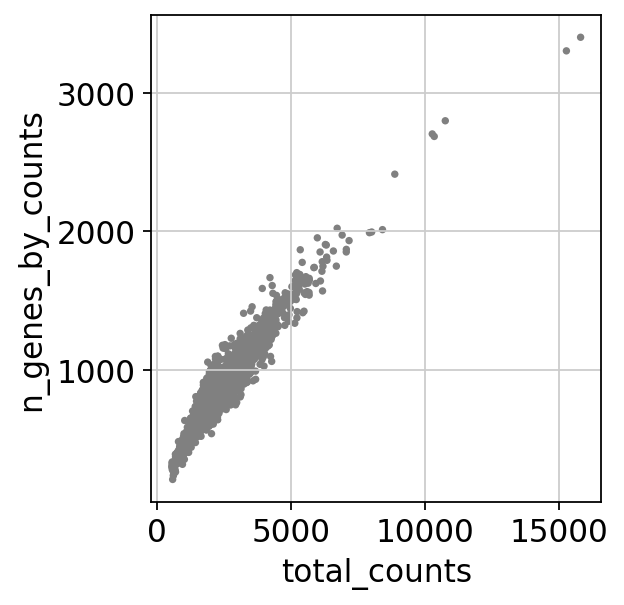
ミトコンドリア由来の遺伝子が多すぎる細胞(5%以上)、またはtotal countsの値が多すぎる細胞(2500以上)を削除します。
[126]:
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :]
adata
[126]:
View of AnnData object with n_obs × n_vars = 2638 × 13714
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts'
2638細胞 x 13714遺伝子のAnnDataになりました。
1.3. 正規化¶
[127]:
# Total read countを10000に正規化
sc.pp.normalize_total(adata, target_sum=1e4)
# 対数変換
sc.pp.log1p(adata)
normalizing counts per cell
finished (0:00:00)
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/preprocessing/_normalization.py:169: UserWarning: Received a view of an AnnData. Making a copy.
view_to_actual(adata)
1.4. highly-variable genesを検出¶
[128]:
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
extracting highly variable genes
finished (0:00:00)
--> added
'highly_variable', boolean vector (adata.var)
'means', float vector (adata.var)
'dispersions', float vector (adata.var)
'dispersions_norm', float vector (adata.var)
1.4.1. highly-variable genesのプロット¶
(左が正規化後、右が正規化前の発現量(Y軸))
[129]:
sc.pl.highly_variable_genes(adata)
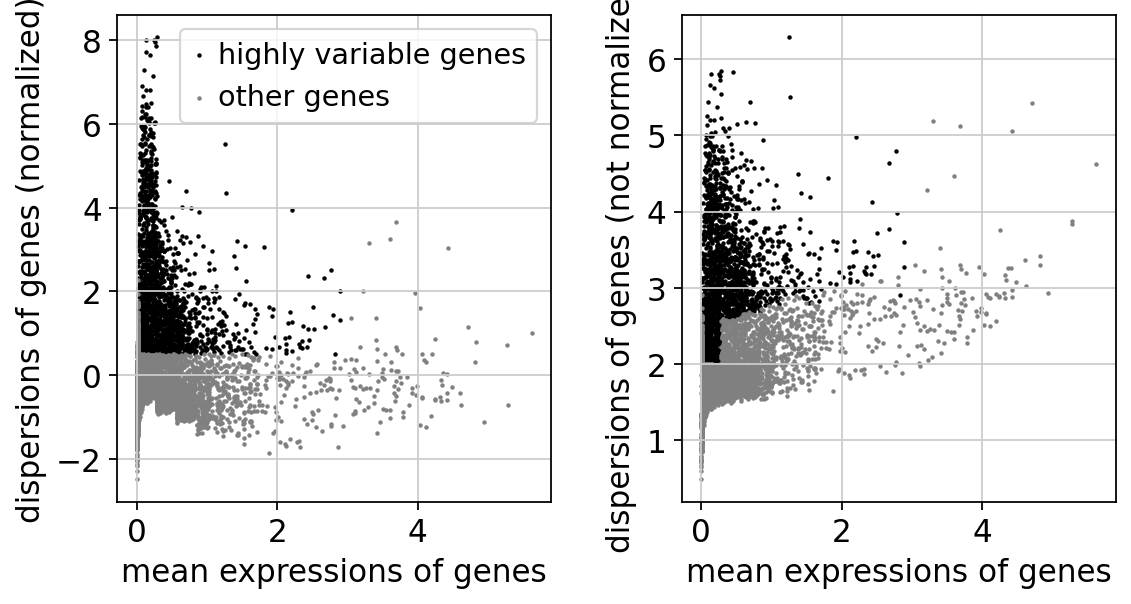
正規化・対数化後の遺伝子発現データをrawdataとして AnnDataオブジェクトの.raw に格納します。
(sc.pp.regress_out を用いたデータ補正と sc.pp.scale を使ったスケーリングを以下で行わない場合は.rawは必要ありません。)
[130]:
adata.raw = adata
以下のコマンドはadataをhighly-variable genesのみに抽出する操作だが、highly-variable genesのリストは .var.highly_variable に保存され、PCAやその後に続く解析では自動的にそれが用いられるため、以下の操作は必要ない。
[131]:
# adata = adata[:, adata.var.highly_variable]
1.4.2. スケーリング¶
細胞あたりのUMI数とミトコンドリア遺伝子の発現率の効果を再調整します。データを単位分散にスケールします。
[132]:
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
# 標準偏差10を超える値はクリップする
sc.pp.scale(adata, max_value=10)
regressing out ['total_counts', 'pct_counts_mt']
sparse input is densified and may lead to high memory use
finished (0:01:01)
1.5. 主成分分析(PCA)¶
[133]:
sc.tl.pca(adata, svd_solver='arpack')
computing PCA
on highly variable genes
with n_comps=50
finished (0:00:00)
[134]:
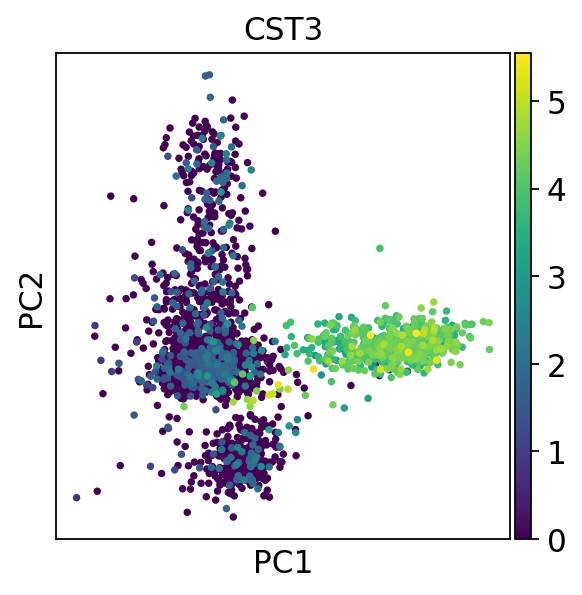
sc.pl.pca(adata, color='CST3')
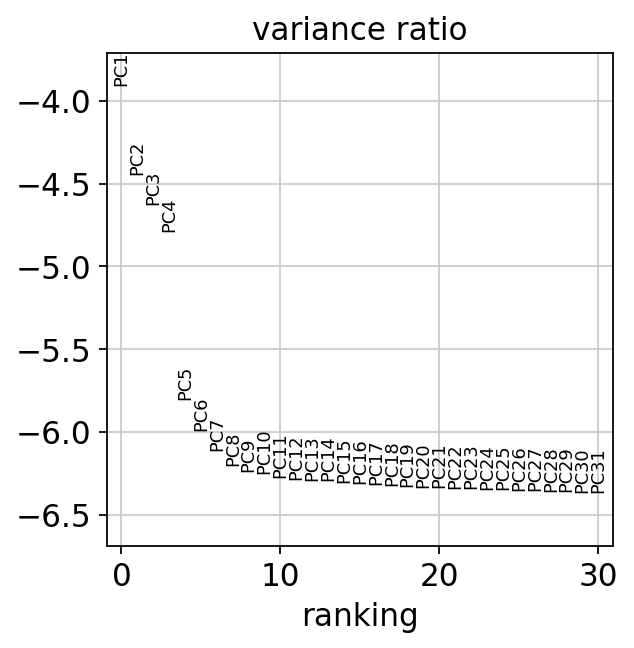
主成分の寄与率を表示します。以降のtSNEやlouvainクラスタリングで使う主成分数をおおまかに見積もります。
[135]:
sc.pl.pca_variance_ratio(adata, log=True)
ここまでの結果をresults_fileに出力
[136]:
adata.write(results_file)
adata
[136]:
AnnData object with n_obs × n_vars = 2638 × 13714
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca'
obsm: 'X_pca'
varm: 'PCs'
1.6. 近傍グラフの計算¶
得られたPCAデータを用いて細胞の近傍グラフを計算します。ここではPC40まで使います。
[137]:
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
computing neighbors
using 'X_pca' with n_pcs = 40
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:00)
1.6.1. 近傍グラフへの埋め込み¶
UMAPを使って2次元まで次元削減します。これはtSNEよりも多様体のグローバルな接続性に忠実である可能性があります。
いくつかのケースでは、クラスタが切断されていたり、似たような連結の切断が見られることがあります。そのような場合には、以下のコメントアウトしている関数を実行してください。
[138]:
#sc.tl.paga(adata)
#sc.pl.paga(adata, plot=False) # remove `plot=False` if you want to see the coarse-grained graph
[139]:
sc.tl.umap(adata)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:03)
1.6.2. 正規化後の発現量でヒートマップ¶
[140]:
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'])
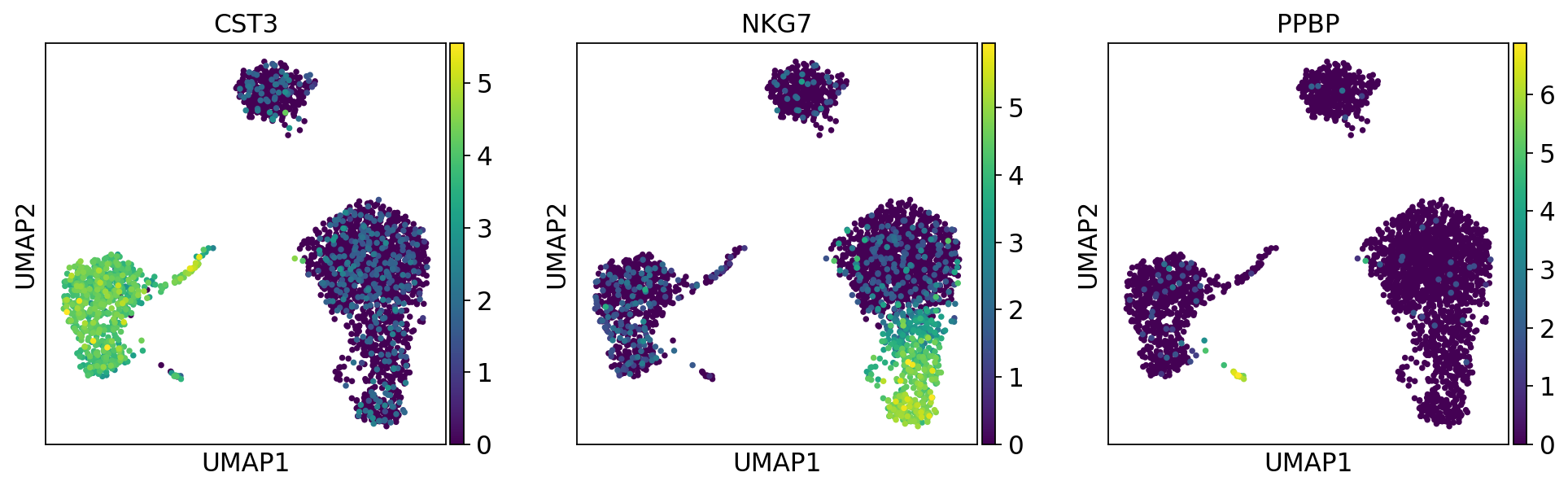
1.6.3. スケーリングした値(Z値)でヒートマップ¶
[141]:
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'], use_raw=False)
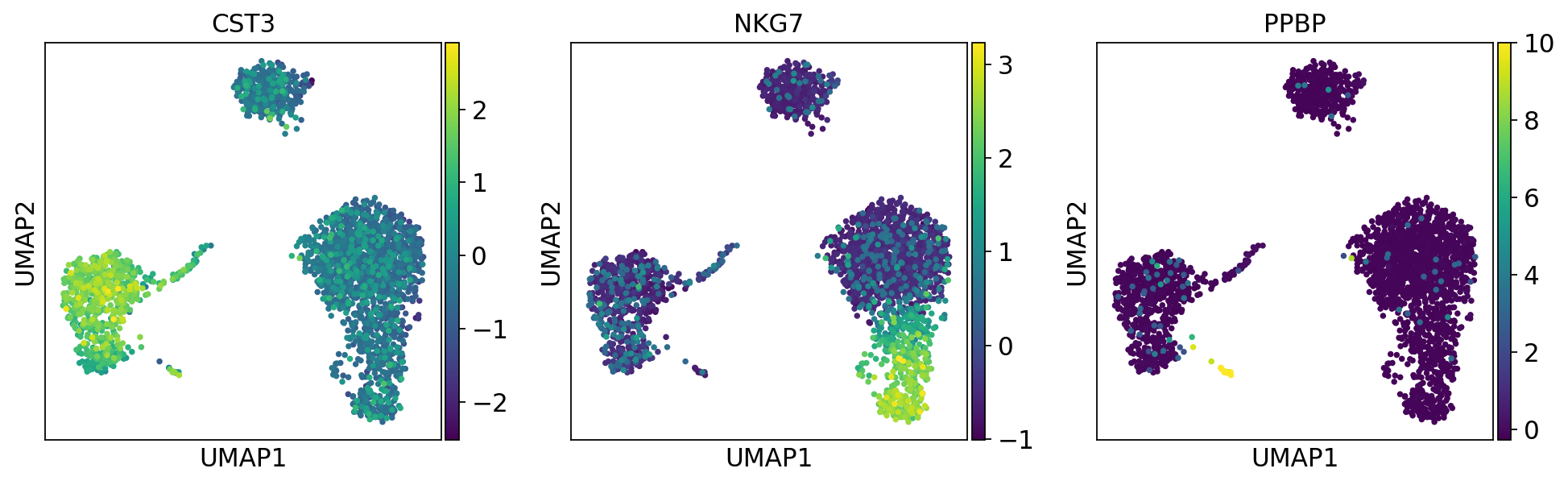
1.7. Clustering¶
他のツール群と同様に、Leiden graph-clustering methodを推奨します。 Leiden clusteringは先ほど計算した neighborhood graphを用いてクラスタリングします。
[142]:
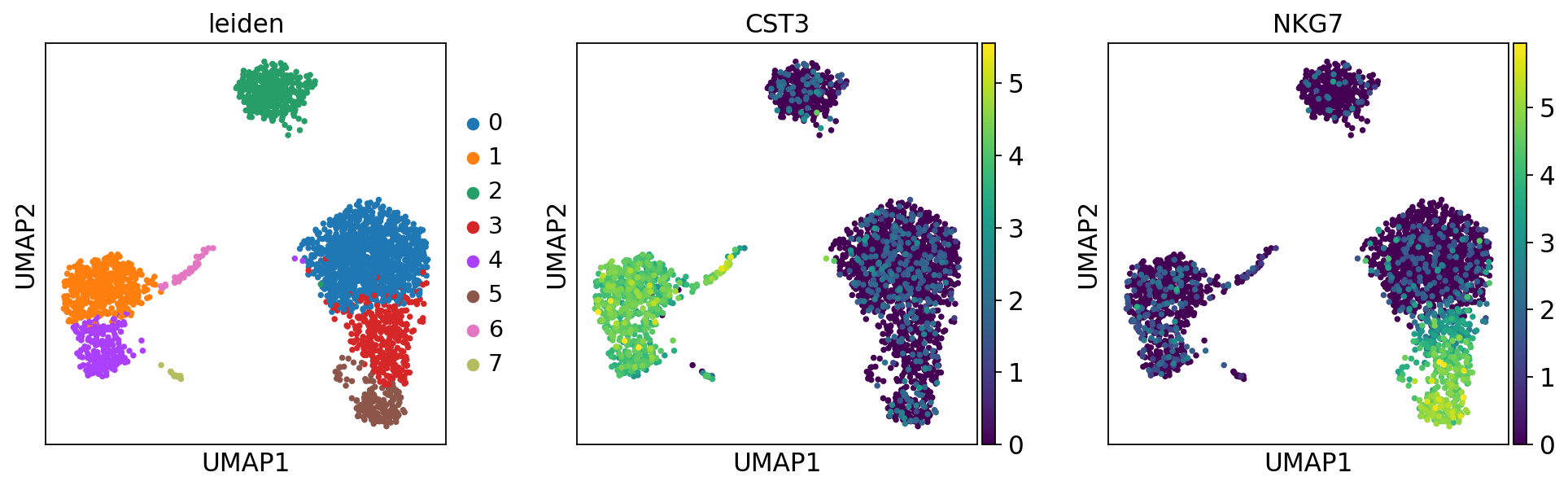
sc.tl.leiden(adata)
running Leiden clustering
finished: found 8 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:00)
[143]:
sc.pl.umap(adata, color=['leiden', 'CST3', 'NKG7'])
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:394: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
[144]:
# データの保存
adata.write(results_file)
1.8. Finding marker genes¶
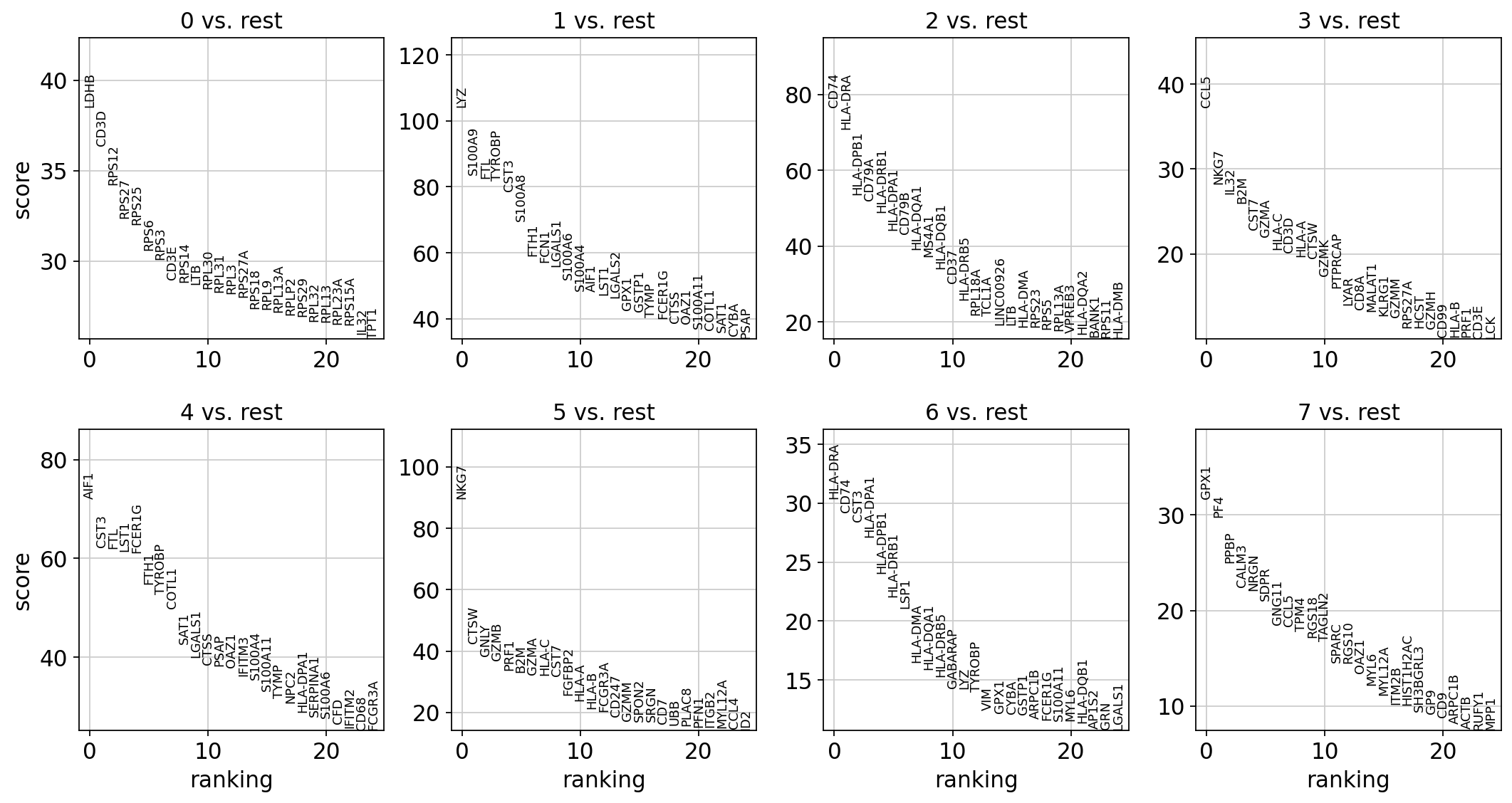
各クラスター内の発現変動遺伝子のランキングを計算します。デフォルトではAnnDataの.raw属性が使用されます。 発現変動解析のための最も単純で最速の方法はt-検定です。
[145]:
# t-test
sc.tl.rank_genes_groups(adata, 'leiden', method='t-test')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
ranking genes
finished: added to `.uns['rank_genes_groups']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:00)
[146]:
sc.settings.verbosity = 2 # reduce the verbosity
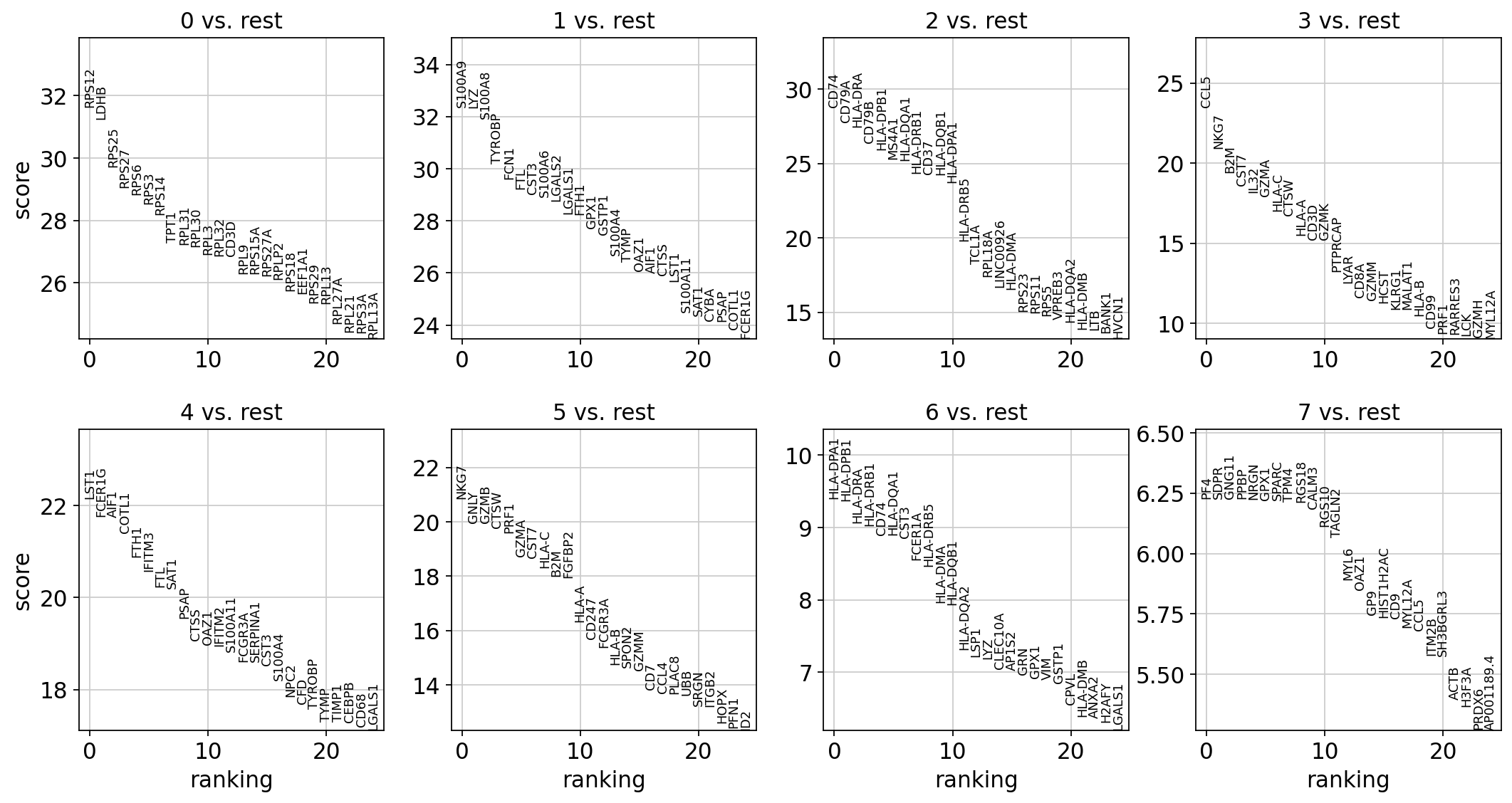
Wilcoxon rank-sum (Mann-Whitney-U) test はt検定と非常に似た結果を返しますが、我々はWilcoxon testを推奨します (see e.g., Sonison & Robinson (2018)).
更に強力な他の手法もあります(例: MAST, limma, DESeq2 and, for python, the recent diffxpy)
[147]:
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
ranking genes
finished (0:00:01)
[148]:
# データの保存
adata.write(results_file)
他の手法として、ロジスティック回帰を用いて遺伝子をランク付けしてみましょう (Natranos et al. (2018))。本質的な違いは、従来の微分検定が一変量であるのに対し、ここでは多変量のアプローチを使用していることです(Clark et al. (2014))。
[149]:
sc.tl.rank_genes_groups(adata, 'leiden', method='logreg')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
ranking genes
finished (0:00:04)
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/sklearn/linear_model/_logistic.py:460: ConvergenceWarning: lbfgs failed to converge (status=1):
STOP: TOTAL NO. of ITERATIONS REACHED LIMIT.
Increase the number of iterations (max_iter) or scale the data as shown in:
https://scikit-learn.org/stable/modules/preprocessing.html
Please also refer to the documentation for alternative solver options:
https://scikit-learn.org/stable/modules/linear_model.html#logistic-regression
n_iter_i = _check_optimize_result(
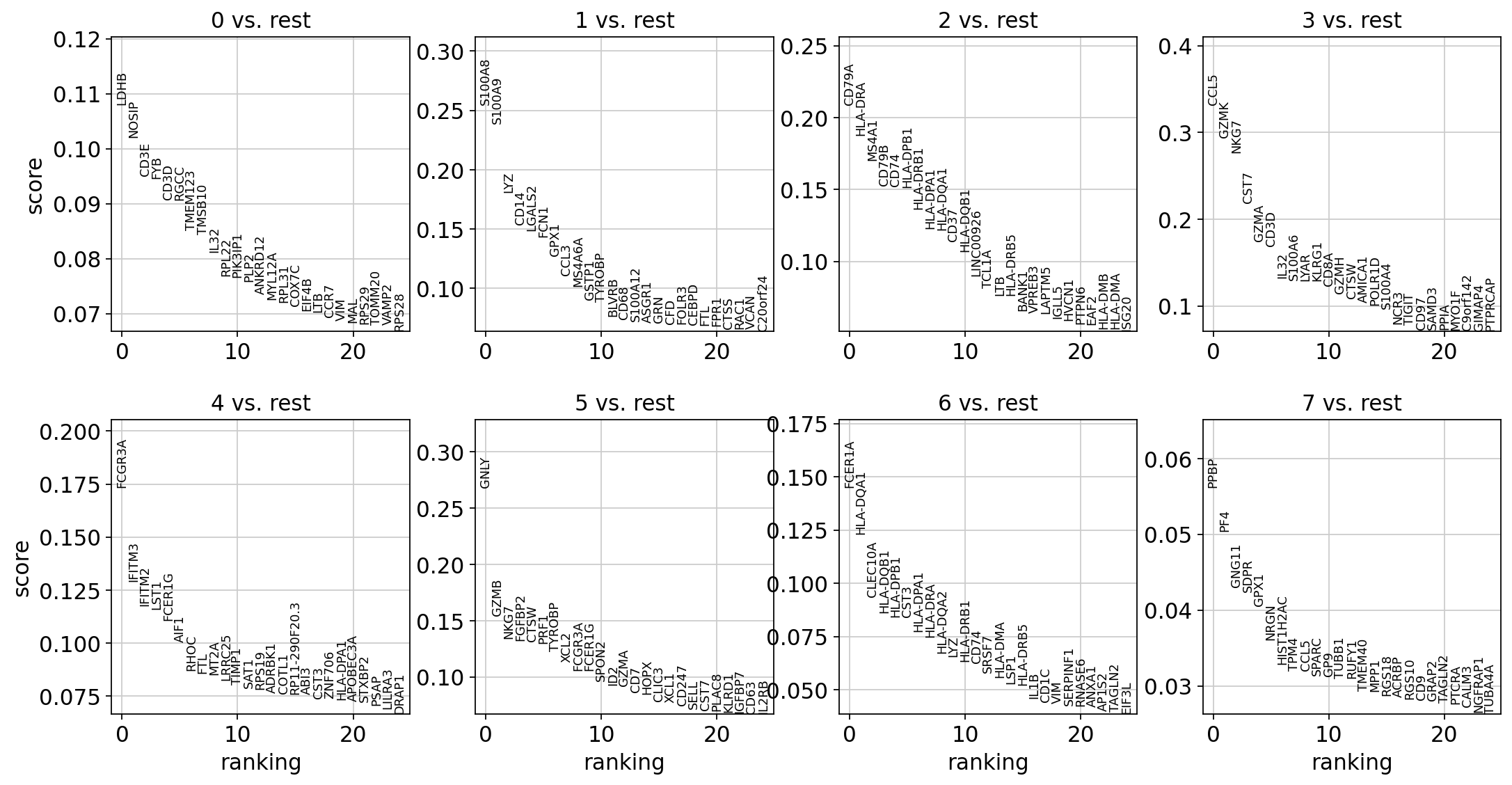
t検定でしか見つからないIL7Rと、他の2つの評価法でしか見つからないFCER1Aを除いて、どの手法でも同様のマーカーを特定できました。
[150]:
# 得られたマーカー遺伝子をリスト化
marker_genes = ['IL7R', 'CD79A', 'MS4A1', 'CD8A', 'CD8B', 'LYZ', 'CD14',
'LGALS3', 'S100A8', 'GNLY', 'NKG7', 'KLRB1',
'FCGR3A', 'MS4A7', 'FCER1A', 'CST3', 'PPBP']
[151]:
# Wilcoxonを用いてやりなおし
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
pd.DataFrame(adata.uns['rank_genes_groups']['names']).head(5)
ranking genes
finished (0:00:02)
[151]:
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|---|
| 0 | RPS12 | S100A9 | CD74 | CCL5 | LST1 | NKG7 | HLA-DPA1 | PF4 |
| 1 | LDHB | LYZ | CD79A | NKG7 | FCER1G | GNLY | HLA-DPB1 | SDPR |
| 2 | RPS25 | S100A8 | HLA-DRA | B2M | AIF1 | GZMB | HLA-DRA | GNG11 |
| 3 | RPS27 | TYROBP | CD79B | CST7 | COTL1 | CTSW | HLA-DRB1 | PPBP |
| 4 | RPS6 | FCN1 | HLA-DPB1 | IL32 | FTH1 | PRF1 | CD74 | NRGN |
[152]:
result = adata.uns['rank_genes_groups']
groups = result['names'].dtype.names
pd.DataFrame(
{group + '_' + key[:1]: result[key][group]
for group in groups for key in ['names', 'pvals']}).head(5)
[152]:
| 0_n | 0_p | 1_n | 1_p | 2_n | 2_p | 3_n | 3_p | 4_n | 4_p | 5_n | 5_p | 6_n | 6_p | 7_n | 7_p | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | RPS12 | 1.402350e-219 | S100A9 | 7.401057e-230 | CD74 | 3.043536e-182 | CCL5 | 3.380869e-122 | LST1 | 9.584233e-109 | NKG7 | 1.203971e-96 | HLA-DPA1 | 5.422417e-21 | PF4 | 4.722886e-10 |
| 1 | LDHB | 2.805350e-214 | LYZ | 2.067780e-229 | CD79A | 6.860832e-170 | NKG7 | 4.248878e-97 | FCER1G | 4.998734e-105 | GNLY | 1.257170e-88 | HLA-DPB1 | 7.591860e-21 | SDPR | 4.733899e-10 |
| 2 | RPS25 | 4.562865e-194 | S100A8 | 9.925222e-224 | HLA-DRA | 8.389292e-166 | B2M | 5.445592e-84 | AIF1 | 6.891419e-105 | GZMB | 1.429027e-88 | HLA-DRA | 1.306768e-19 | GNG11 | 4.733899e-10 |
| 3 | RPS27 | 1.164214e-185 | TYROBP | 1.541222e-200 | CD79B | 1.171444e-153 | CST7 | 1.850639e-77 | COTL1 | 1.329234e-101 | CTSW | 4.144726e-87 | HLA-DRB1 | 1.865104e-19 | PPBP | 4.744938e-10 |
| 4 | RPS6 | 8.893675e-183 | FCN1 | 9.951201e-193 | HLA-DPB1 | 6.167786e-148 | IL32 | 7.057861e-74 | FTH1 | 8.135658e-97 | PRF1 | 1.692100e-85 | CD74 | 5.853161e-19 | NRGN | 4.800511e-10 |
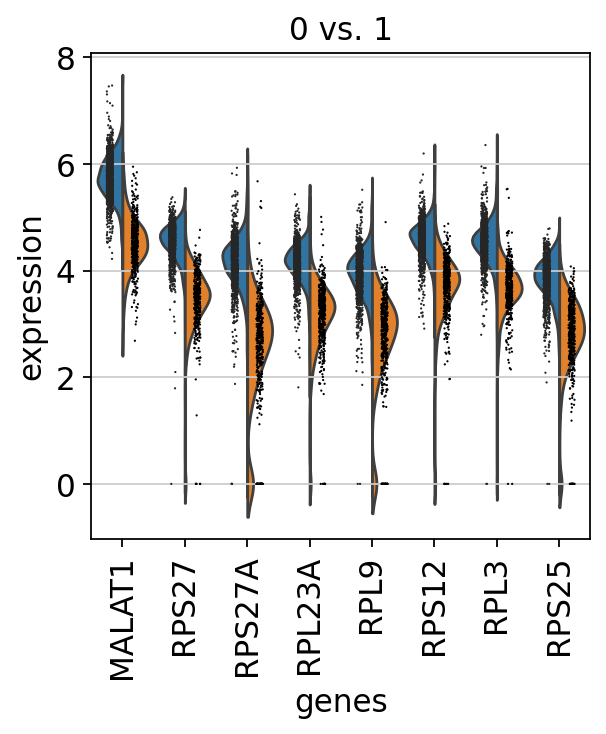
1.8.1. クラスター0と1を比較¶
[153]:
# DEGランキング
sc.tl.rank_genes_groups(adata, 'leiden', groups=['0'], reference='1', method='wilcoxon')
sc.pl.rank_genes_groups(adata, groups=['0'], n_genes=20)
#発現量のviolin plot
sc.pl.rank_genes_groups_violin(adata, groups='0', n_genes=8)
ranking genes
finished (0:00:00)
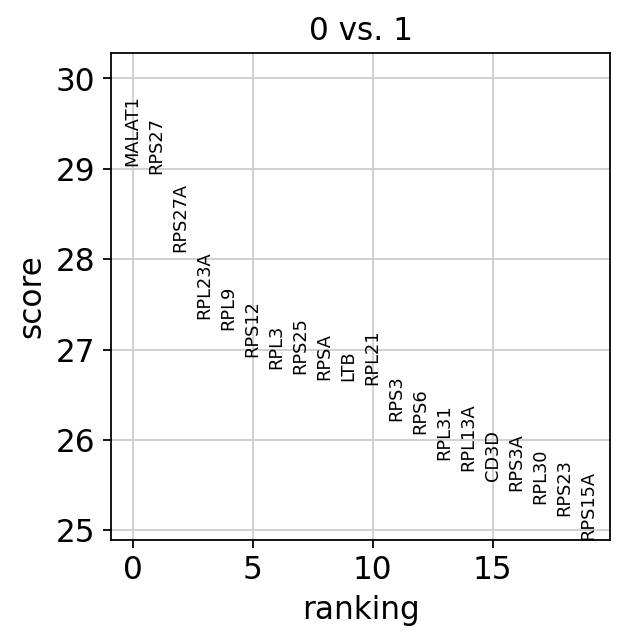
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_tools/__init__.py:1170: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_tools/__init__.py:1183: FutureWarning:
Setting a gradient palette using color= is deprecated and will be removed in v0.14.0. Set `palette='dark:black'` for the same effect.
_ax = sns.stripplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_tools/__init__.py:1199: UserWarning: FixedFormatter should only be used together with FixedLocator
_ax.set_xticklabels(new_gene_names, rotation='vertical')
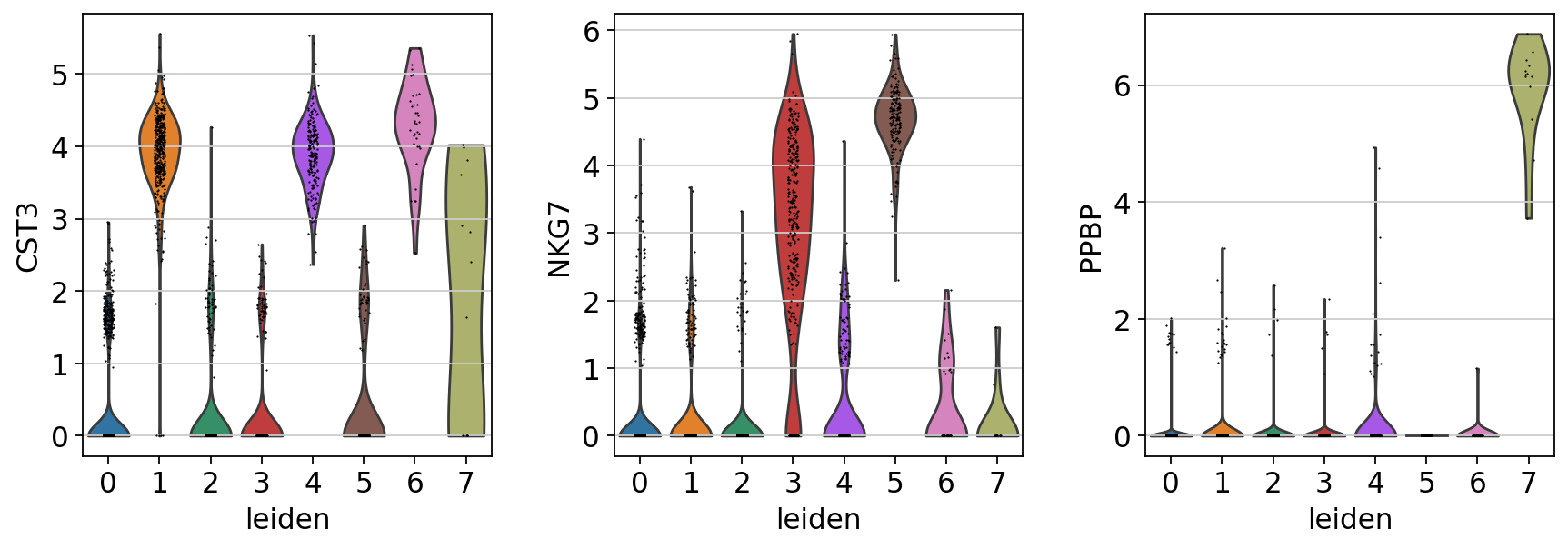
1.8.2. 1対他クラスターの比較¶
[154]:
adata = sc.read(results_file)
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
sc.pl.rank_genes_groups_violin(adata, groups='0', n_genes=8)
ranking genes
finished (0:00:02)
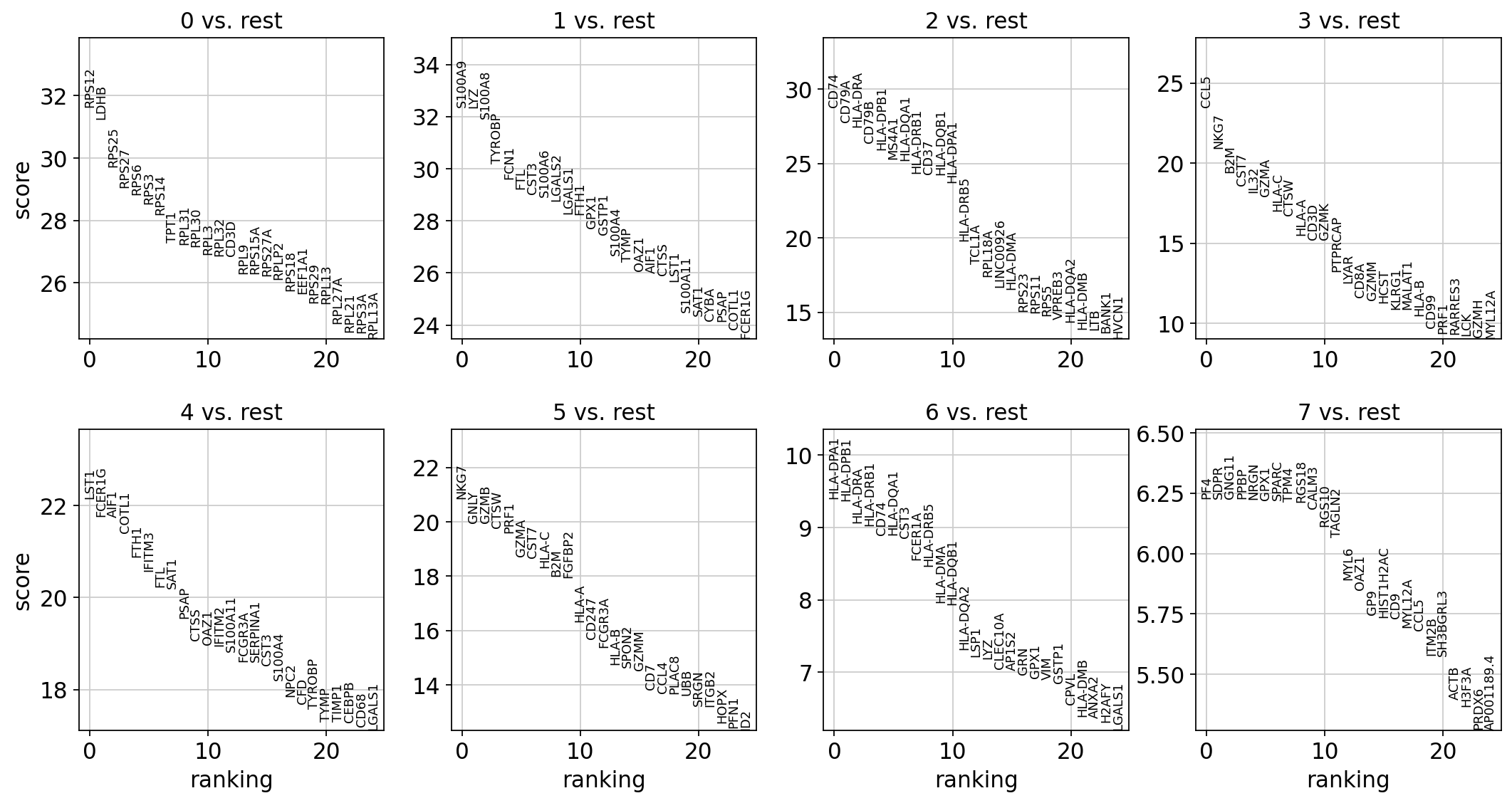
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_tools/__init__.py:1170: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_tools/__init__.py:1183: FutureWarning:
Setting a gradient palette using color= is deprecated and will be removed in v0.14.0. Set `palette='dark:black'` for the same effect.
_ax = sns.stripplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_tools/__init__.py:1199: UserWarning: FixedFormatter should only be used together with FixedLocator
_ax.set_xticklabels(new_gene_names, rotation='vertical')
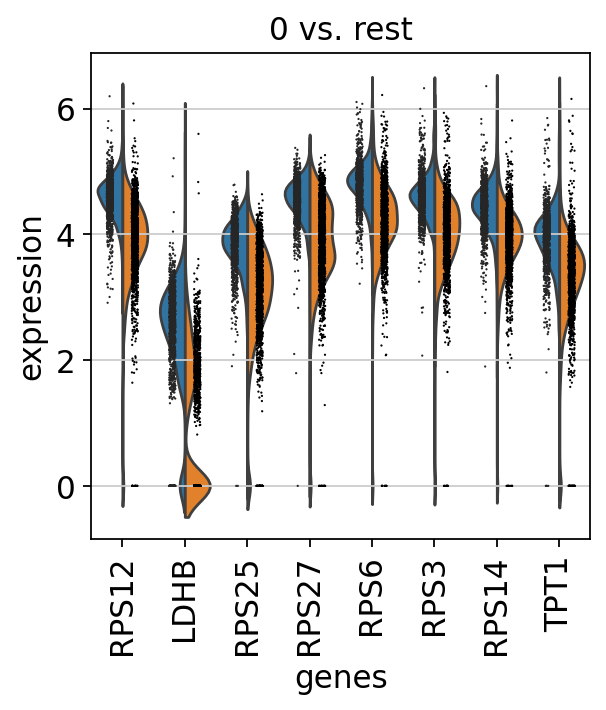
[155]:
# 指定した遺伝子のクラスター間発現量比較
sc.pl.violin(adata, ['CST3', 'NKG7', 'PPBP'], groupby='leiden')
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
ax = sns.violinplot(
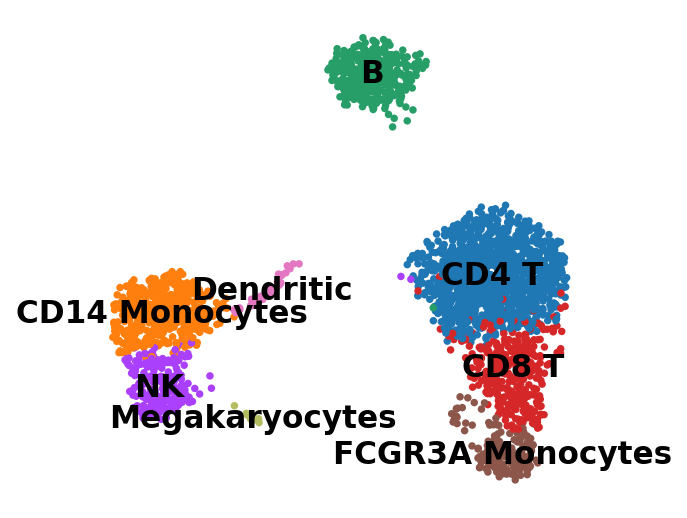
1.8.3. クラスターにラベルを付与¶
[156]:
new_cluster_names = [
'CD4 T',
'CD14 Monocytes',
'B',
'CD8 T',
'NK',
'FCGR3A Monocytes',
'Dendritic',
'Megakaryocytes']
#adata.rename_categories('leiden', new_cluster_names) エラーが出るため暫定的に以下のようにする
adata.obs['leiden'] = adata.obs['leiden'].cat.rename_categories(new_cluster_names)
[157]:
sc.pl.umap(adata, color='leiden', legend_loc='on data', title='', frameon=False, save='.pdf')
WARNING: saving figure to file figures/umap.pdf
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:394: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
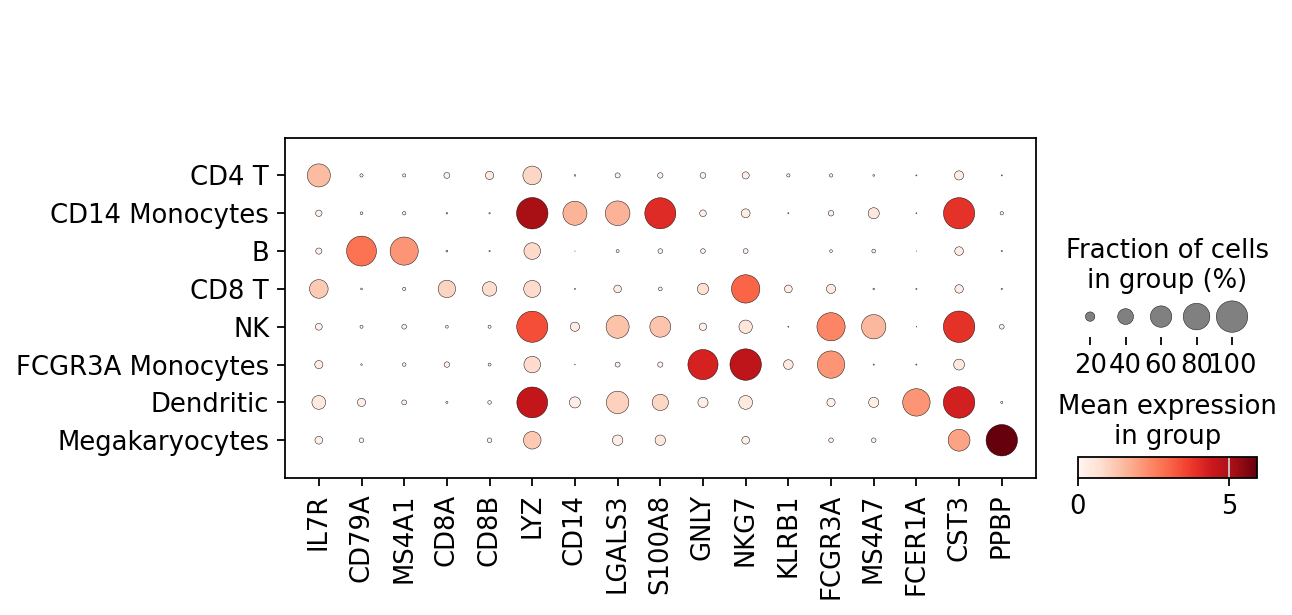
marker_genes を使ってマーカー遺伝子の発現量ヒートマップ
(作成者注:上のviolin plotでは分布がわかるが数遺伝子しか可視化できないのに対し、下のヒートマップでは平均値のみの表示だが多数の遺伝子を同時表示可能。)
[158]:
sc.pl.dotplot(adata, marker_genes, groupby='leiden');
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_dotplot.py:747: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap', 'norm' will be ignored
dot_ax.scatter(x, y, **kwds)
violin plotにすることも可能
(作成者注:busyなのであまりおすすめしない)
[159]:
sc.pl.stacked_violin(adata, marker_genes, groupby='leiden') # , rotation=90
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: UserWarning: Numpy array is not a supported type for `palette`. Please convert your palette to a list. This will become an error in v0.14
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: UserWarning: Numpy array is not a supported type for `palette`. Please convert your palette to a list. This will become an error in v0.14
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: UserWarning: Numpy array is not a supported type for `palette`. Please convert your palette to a list. This will become an error in v0.14
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: UserWarning: Numpy array is not a supported type for `palette`. Please convert your palette to a list. This will become an error in v0.14
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: UserWarning: Numpy array is not a supported type for `palette`. Please convert your palette to a list. This will become an error in v0.14
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: UserWarning: Numpy array is not a supported type for `palette`. Please convert your palette to a list. This will become an error in v0.14
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: UserWarning: Numpy array is not a supported type for `palette`. Please convert your palette to a list. This will become an error in v0.14
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: UserWarning: Numpy array is not a supported type for `palette`. Please convert your palette to a list. This will become an error in v0.14
row_ax = sns.violinplot(
/opt/conda/envs/shortcake_default/lib/python3.8/site-packages/scanpy/plotting/_stacked_violin.py:461: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
row_ax = sns.violinplot(
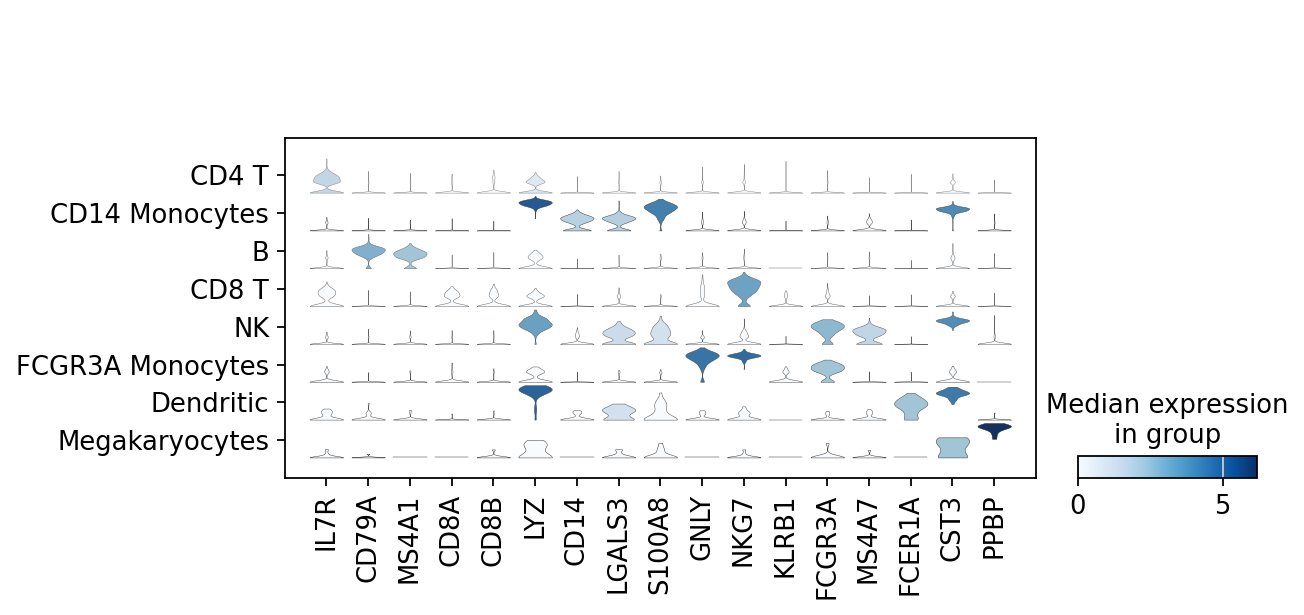
この分析の中で取得されたアノテーションを表示
[160]:
adata
[160]:
AnnData object with n_obs × n_vars = 2638 × 13714
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'leiden'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'hvg', 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'rank_genes_groups', 'umap'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'connectivities', 'distances'
compression='gzip' で圧縮形式でオブジェクト保存
[161]:
adata.write(results_file, compression='gzip')
保存した
.h5adファイルは、h5lsコマンド を使ってファイルの大まかな概要を知ることができますこのファイルフォーマットは将来的にさらに最適化される可能性がありますが、すべての読み込み機能は後方互換性があります。
[162]:
!h5ls ./write/pbmc3k.h5ad
X Dataset {2638, 13714}
layers Group
obs Group
obsm Group
obsp Group
raw Group
uns Group
var Group
varm Group
varp Group
このオブジェクトを単に可視化のために使いたい人と共有したい場合、ファイルサイズを小さくする簡単な方法は、スケーリング/補正された発現行列を削除することです。
下記コマンドで
adata.rawに保存された raw dataを保存することができます。
[163]:
adata.raw.to_adata().write('./write/pbmc3k_withoutX.h5ad')
csv形式で保存したい場合は以下のようにします:
[164]:
# Export single fields of the annotation of observations
# adata.obs[['n_counts', 'louvain_groups']].to_csv(
# './write/pbmc3k_corrected_louvain_groups.csv')
# Export single columns of the multidimensional annotation
# adata.obsm.to_df()[['X_pca1', 'X_pca2']].to_csv(
# './write/pbmc3k_corrected_X_pca.csv')
# Or export everything except the data using `.write_csvs`.
# Set `skip_data=False` if you also want to export the data.
# adata.write_csvs(results_file[:-5], )
[165]:
import session_info
session_info.show()
[165]:
Click to view session information
----- anndata 0.8.0 numpy 1.24.3 pandas 2.0.3 scanpy 1.9.8 session_info 1.0.0 -----
Click to view modules imported as dependencies
PIL 10.0.1 anyio NA asciitree NA asttokens NA attr 23.1.0 attrs 23.1.0 babel 2.11.0 backcall 0.2.0 bottleneck 1.3.5 brotli 1.0.9 certifi 2024.02.02 cffi 1.16.0 charset_normalizer 2.0.4 cloudpickle 3.0.0 colorama 0.4.6 comm 0.1.2 cycler 0.10.0 cython_runtime NA dask 2023.5.0 dateutil 2.8.2 debugpy 1.6.7 decorator 5.1.1 defusedxml 0.7.1 entrypoints 0.4 executing 0.8.3 fasteners 0.19 fastjsonschema NA fontTools 4.25.0 h5py 3.8.0 idna 3.4 igraph 0.11.3 importlib_metadata NA importlib_resources NA ipykernel 6.25.0 ipython_genutils 0.2.0 ipywidgets 7.7.1 jedi 0.18.1 jinja2 3.1.3 joblib 1.3.2 json5 NA jsonschema 4.19.2 jsonschema_specifications NA jupyter_server 1.23.4 jupyterlab_server 2.25.1 kiwisolver 1.4.4 leidenalg 0.10.2 llvmlite 0.41.1 louvain 0.8.1 lxml 5.1.0 markupsafe 2.1.3 matplotlib 3.7.2 matplotlib_inline 0.1.6 mkl 2.4.0 mpl_toolkits NA natsort 8.4.0 nbformat 5.9.2 numba 0.58.1 numcodecs 0.12.1 numexpr 2.8.4 packaging 23.1 parso 0.8.3 patsy 0.5.6 pexpect 4.8.0 pickleshare 0.7.5 pkg_resources NA platformdirs 3.10.0 plotly 5.18.0 prometheus_client NA prompt_toolkit 3.0.43 psutil 5.9.0 ptyprocess 0.7.0 pure_eval 0.2.2 pyarrow 15.0.0 pycparser 2.21 pydev_ipython NA pydevconsole NA pydevd 2.9.5 pydevd_file_utils NA pydevd_plugins NA pydevd_tracing NA pygments 2.15.1 pynndescent 0.5.11 pyparsing 3.0.9 pytz 2023.3.post1 referencing NA requests 2.31.0 rfc3339_validator 0.1.4 rfc3986_validator 0.1.1 rpds NA scipy 1.10.1 seaborn 0.13.2 send2trash NA setuptools 68.2.2 setuptools_scm NA six 1.16.0 sklearn 1.3.2 sniffio 1.3.0 socks 1.7.1 stack_data 0.2.0 statsmodels 0.14.1 terminado 0.17.1 texttable 1.7.0 threadpoolctl 3.2.0 tlz 0.12.1 tomli 2.0.1 toolz 0.12.1 tornado 6.3.3 tqdm 4.66.1 traitlets 5.7.1 typing_extensions NA umap 0.5.5 urllib3 1.26.18 wcwidth 0.2.5 websocket 0.58.0 yaml 6.0.1 zarr 2.16.1 zipp NA zmq 23.2.0 zope NA
----- IPython 8.12.2 jupyter_client 7.4.9 jupyter_core 5.5.0 jupyterlab 3.6.3 notebook 6.5.4 ----- Python 3.8.18 (default, Sep 11 2023, 13:40:15) [GCC 11.2.0] Linux-5.15.0-91-generic-x86_64-with-glibc2.17 ----- Session information updated at 2024-02-06 21:44